This project focuses on human population genomics. We infer human demographic history, such as global migrations, population size changes, and mixing between populations through modeling. This type of research satisfies an innate human interest to understand our own history, and provides a foundation for medical genomics research.
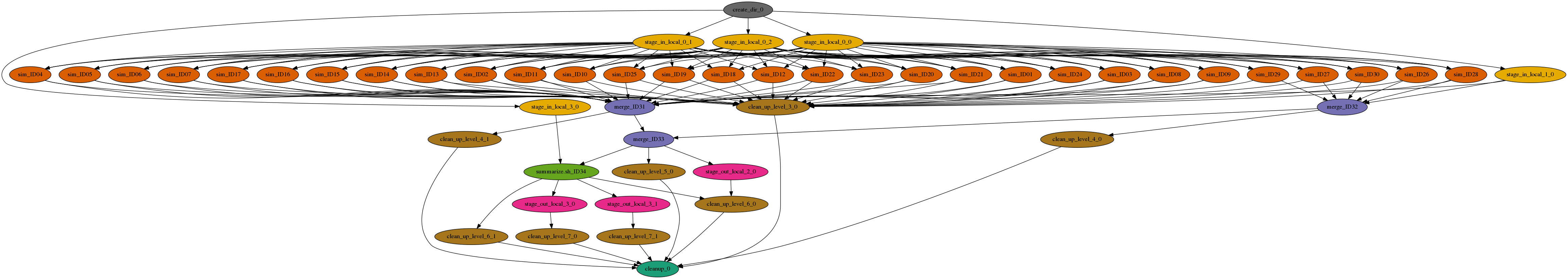
Specifically, in this project we infer the demographic history of Ashkenazi Jews. We use Approximate Bayesian Computation to choose the best model of Ashkenazi history and then estimate the parameter values for the best model. First, 1 million independent simulations are run for each demographic model, then genomic summary statistics are calculated using the simulated genomes and real data, and finally the model and parameters that give the closest statistics to the real data are identified. The Pegasus workflow completes the first two steps, and the final step is done separately. The workflow is a large set of independent simulations (100,000s to millions), followed by a set of merge and data summarize tasks.
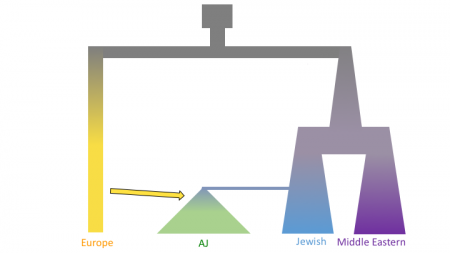
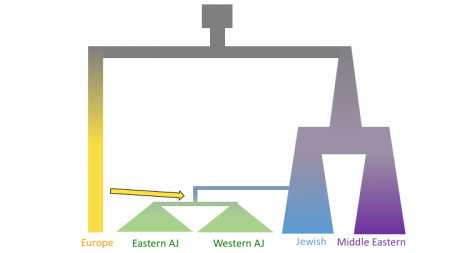
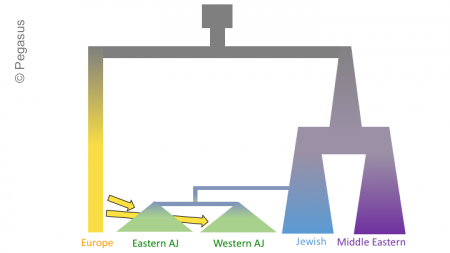
The figures below shows three models of Ashkenazi demographic history. Time is on the y-axis and effective population size is on the x-axis. The past is at the top, and the present is at that bottom. These models share and old population size reduction (Out of Africa), population split between the ancestors of populations in Europe and the Middle East, exponential population growth in the Jewish populations and populations from the Middle East, a population split between Jewish populations and populations from the Middle East, a founder event in the Ashkenazi Jews, and gene flow from Europeans to the Ashkenazi Jews. In the second and third models there is an additional population split within the Ashkenazi Jews, and in the third model there is the possibility of different amounts of gene flow from the Europeans to the two Ashkenazi groups.
Researchers: Ariella Gladstein (agladstein@email.arizona.edu) and Michael Hammer, Univeristy of Arizona