12. Optimizing Workflows for Efficiency and Scalability
By default, Pegasus generates workflows which targets the most common use cases and execution environments. For more specialized environments or workflows, the following sections can provide hints on how to optimize your workflow to scale better, and run more efficiently. Below are some common issues and solutions.
12.1. Optimizing Short Jobs / Scheduling Delays
Issue: Even though HTCondor is a high throughput system, there are overheads when scheduling short jobs. Common overheads include scheduling, data transfers, state notifications, and task book keeping. These overheads can be very noticeable for short jobs, but not noticeable at all for longer jobs as the ratio between the computation and the overhead is higher.
Solution: If you have many short jobs to run, the solution to minimize the overheads is to use job clustering. This instructs Pegasus to take a set of jobs, selected horizontally, by labels, or by runtime, and create jobs containing that whole set of tasks. The result is more efficient jobs, for which the overheads are less noticeable.
Tip
We generally recommend that your jobs should run at least 10 minutes, to make the various delays worthwhile. That is a good ballpark to keep in mind when clustering short running tasks.
12.2. Job Clustering
A large number of workflows executed through the Pegasus Workflow Management System are composed of several jobs that run for only a few seconds or so. The overhead of running any job on the grid is usually 60 seconds or more. Hence, it makes sense to cluster small independent jobs into a larger job. This is done while mapping an abstract workflow to an executable workflow. Site specific or transformation specific criteria are taken into consideration while clustering smaller jobs into a larger job in the executable workflow. The user is allowed to control the granularity of this clustering on a per transformation, per site basis.
12.2.1. Overview
The abstract workflow is mapped onto the various sites by the Site Selector. This semi executable workflow is then passed to the clustering module. The clustering of the workflow can be either be:
level based horizontal clustering - where you can denote how many jobs get clustered into a single clustered job per level, or how many clustered jobs should be created per level of the workflow
level based runtime clustering - similar to horizontal clustering, but while creating the clusters per level take into account the job runtimes.
label based (clustering by label)
The clustering module clusters the jobs into larger/clustered jobs, that
can then be executed on the remote sites. The execution can either be
sequential on a single node, or on multiple nodes using MPI. To specify
which clustering technique to use the user has to pass the --cluster
option to pegasus-plan.
12.2.2. Generating Clustered Executable Workflow
The clustering of a workflow is activated by passing the
--cluster | -C option to pegasus-plan. The clustering
granularity of a particular logical transformation on a particular site is dependant
upon the clustering techniques being used. The executable that is used
for running the clustered job on a particular site is determined as
explained in section 7.
#Running pegasus-plan to generate clustered workflows
$ pegasus-plan --dir ./dags -p siteX --output local
--cluster [comma separated list of clustering techniques] workflow.yml
Valid clustering techniques are "horizontal" and "label".
The naming convention of submit files of the clustered jobs
is merge_NAME_IDX.sub . The NAME is derived from the logical
transformation name. The IDX is an integer number between 1 and the
total number of jobs in a cluster. Each of the submit files has a
corresponding input file, following the naming convention
merge_NAME_IDX.in. The input file contains the respective execution
targets and the arguments for each of the jobs that make up the
clustered job.
12.2.3. Horizontal Clustering
In case of horizontal clustering, each job in the workflow is associated
with a level. The levels of the workflow are determined by doing a
modified Breadth First Traversal of the workflow starting from the root
nodes. The level associated with a node, is the furthest distance of it
from the root node instead of it being the shortest distance as in
normal BFS. For each level the jobs are grouped by the site on which
they have been scheduled by the Site Selector. Only jobs of same type
(txnamespace, txname, txversion) can be clustered into a larger job. To
use horizontal clustering the user needs to set the --cluster option
of pegasus-plan to horizontal.
12.2.3.1. Controlling Clustering Granularity
The number of jobs that have to be clustered into a single large job, is
determined by the value of two parameters associated with the smaller
jobs. Both these parameters are specified by the use of PEGASUS
namespace profile keys. The keys can be specified at any of the
placeholders for the profiles (abstract transformation in the Abstract Workflow, site
in the site catalog, transformation in the transformation catalog). The
normal overloading semantics apply i.e. profile in transformation
catalog overrides the one in the site catalog and that in turn overrides
the one in the Abstract Workflow. The two parameters are described below.
clusters.size factor
The
clusters.sizefactor denotes how many jobs need to be merged into a single clustered job. It is specified via the use of aPEGASUSnamespace profile keyclusters.size. For e.g. if at a particular level, say 4 jobs referring to logical transformation B have been scheduled to a siteX. Theclusters.sizefactor associated with job B for siteX is say 3. This will result in 2 clustered jobs, one composed of 3 jobs and another of 1 job. Theclusters.sizefactor can be specified in the transformation catalog as follows#!/usr/bin/env python3 from Pegasus.api import * # create the TransformationCatalog object tc = TransformationCatalog() # create and add the transformation B = Transformation( "B", site="siteX", pfn="/shared/PEGASUS/bin/jobB", is_stageable=False, ).add_profiles(Namespace.PEGASUS, key="clusters.size", value=3) tc.add_transformations(B) C = Transformation( "C", site="siteX", pfn="/shared/PEGASUS/bin/jobC", is_stageable=False, ).add_profiles(Namespace.PEGASUS, key="clusters.size" value=2) tc.add_transformations(C) # write the transformation catalog to the default file path "./transformations.yml" tc.write()
x-pegasus: {apiLang: python, createdBy: vahi, createdOn: '10-29-20T13:39:30Z'} pegasus: '5.0' transformations: - name: B sites: - {name: siteX, pfn: /shared/PEGASUS/bin/jobB, type: installed} profiles: pegasus: {clusters_size: 3} - name: C sites: - {name: siteX, pfn: /shared/PEGASUS/bin/jobC, type: installed} profiles: pegasus: {clusters_size: 2}
# multiple line text-based transformation catalog: 2014-09-30T16:05:01.731-07:00 tr B { site siteX { profile pegasus "clusters.size" "3" pfn "/shared/PEGASUS/bin/jobB" arch "x86" os "LINUX" type "INSTALLED" } } tr C { site siteX { profile pegasus "clusters.size" "2" pfn "/shared/PEGASUS/bin/jobC" arch "x86" os "LINUX" type "INSTALLED" } }
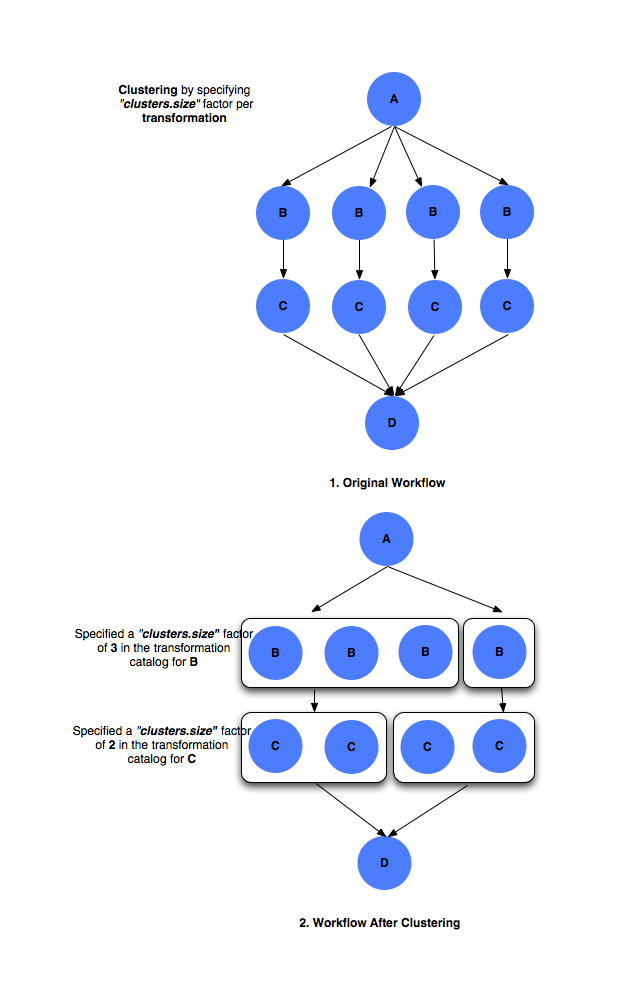
Clustering by
clusters.sizeclusters.num factor
The
clusters.numfactor denotes how many clustered jobs does the user want to see per level per site. It is specified via the use of aPEGASUSnamespace profile keyclusters.num. for e.g. if at a particular level, say 4 jobs referring to logical transformation B have been scheduled to a siteX. Theclusters.numfactor associated with job B for siteX is say 3. This will result in 3 clustered jobs, one composed of 2 jobs and others of a single job each. Theclusters.numfactor in the transformation catalog can be specified as follows#!/usr/bin/env python3 from Pegasus.api import * # create the TransformationCatalog object tc = TransformationCatalog() # create and add the transformation B = Transformation( "B", site="siteX", pfn="/shared/PEGASUS/bin/jobB", is_stageable=False, ).add_profiles(Namespace.PEGASUS, key="clusters.num", value=3) tc.add_transformations(B) C = Transformation( "C", site="siteX", pfn="/shared/PEGASUS/bin/jobC", is_stageable=False, ).add_profiles(Namespace.PEGASUS, key="clusters.num", value=2) tc.add_transformations(C) # write the transformation catalog to the default file path "./transformations.yml" tc.write()
x-pegasus: {apiLang: python, createdBy: vahi, createdOn: '10-29-20T13:39:30Z'} pegasus: '5.0' transformations: - name: B sites: - {name: siteX, pfn: /shared/PEGASUS/bin/jobB, type: installed} profiles: pegasus: {clusters.num: 3} - name: C sites: - {name: siteX, pfn: /shared/PEGASUS/bin/jobC, type: installed} profiles: pegasus: {clusters.num: 2}
# multiple line text-based transformation catalog: 2014-09-30T16:05:01.731-07:00 tr B { site siteX { profile pegasus "clusters.num" "3" pfn "/shared/PEGASUS/bin/jobB" arch "x86" os "LINUX" type "INSTALLED" } } tr C { site siteX { profile pegasus "clusters.num" "2" pfn "/shared/PEGASUS/bin/jobC" arch "x86" os "LINUX" type "INSTALLED" } }
In the case, where both the factors are associated with the job, the
clusters.numvalue supersedes theclusters.sizevalue.#!/usr/bin/env python3 from Pegasus.api import * # create the TransformationCatalog object tc = TransformationCatalog() # create and add the transformation B = Transformation( "B", site="siteX", pfn="/shared/PEGASUS/bin/jobB", is_stageable=False, ).add_pegasus_profiles(clusters_num=3, clusters_size=3) tc.add_transformations(B) # write the transformation catalog to the default file path "./transformations.yml" tc.write()
x-pegasus: {apiLang: python, createdBy: vahi, createdOn: '10-29-20T13:39:30Z'} pegasus: '5.0' transformations: - name: B sites: - {name: siteX, pfn: /shared/PEGASUS/bin/jobB, type: installed} profiles: pegasus: {clusters.num: 3, clusters.size:3}
# multiple line text-based transformation catalog: 2014-09-30T16:05:01.731-07:00 tr B { site siteX { profile pegasus "clusters.num" "3" profile pegasus "clusters.size" "3" pfn "/shared/PEGASUS/bin/jobB" arch "x86" os "LINUX" type "INSTALLED" } }
In the above case the jobs referring to logical transformation B scheduled on siteX will be clustered on the basis of
clusters.numvalue. Hence, if there are 4 jobs referring to logical transformation B scheduled to siteX, then 3 clustered jobs will be created.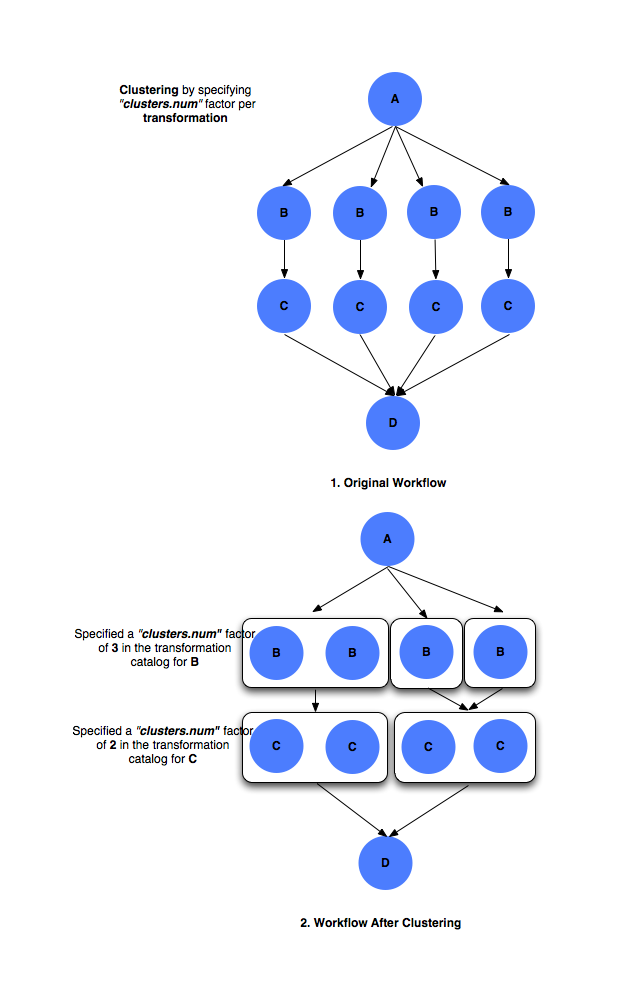
Clustering by
clusters.num
12.2.4. Runtime Clustering
Workflows often consist of jobs of same type, but have varying run times. Two or more instances of the same job, with varying inputs can differ significantly in their runtimes. A simple way to think about this is running the same program on two distinct input sets, where one input is smaller, say 1 MB, as compared to the other which is 10 GB in size. In such a case the two jobs will have significantly differing run times. When such jobs are clustered using horizontal clustering, the benefits of job clustering may be lost if all smaller jobs get clustered together, while the larger jobs are clustered together. In such scenarios it would be beneficial to be able to cluster jobs together such that all clustered jobs have similar runtimes.
In case of runtime clustering, jobs in the workflow are associated with
a level. The levels of the workflow are determined in the same manner as
in horizontal clustering. For each level the jobs are grouped by the
site on which they have been scheduled by the Site Selector. Only jobs
of same type (txnamespace, txname, txversion) can be clustered into a
larger job. To use runtime clustering the user needs to set the
--cluster option of pegasus-plan horizontal, and set the
Pegasus property pegasus.clusterer.preference to Runtime.
Runtime clustering supports two modes of operation.
Clusters jobs together such that the clustered job’s runtime does not exceed a user specified maxruntime.
Basic Algorithm of grouping jobs into clusters is as follows
// cluster.maxruntime - Is the maximum runtime for which the clustered job should run. // j.runtime - Is the runtime of the job j. 1. Create a set of jobs of the same type (txnamespace, txname, txversion), and that run on the same site. 2. Sort the jobs in decreasing order of their runtime. 3. For each job j, repeat a. If j.runtime > cluster.maxruntime then ignore j. // Sum of runtime of jobs already in the bin + j.runtime <= cluster.maxruntime b. If j can be added to any existing bin (clustered job) then Add j to bin Else Add a new bin Add job j to newly added bin
The runtime of a job, and the maximum runtime for which a clustered job should run is determined by the value of two parameters associated with the jobs.
runtime
expected runtime for a job
clusters.maxruntime
maxruntime for the clustered job i.e. Group as many jobs as possible into a cluster, as long as the clustered jobs’ runtime does not exceed clusters.maxruntime.
Clusters all the jobs into a fixed number of clusters (
clusters.num), such that the runtimes of the clustered jobs are similar.Basic Algorithm of grouping jobs into clusters is as follows
// cluster.num - Is the number of clustered jobs to create. // j.runtime - Is the runtime of the job j. 1. Create a set of jobs of the same type (txnamespace, txname, txversion), and that run on the same site. 2. Sort the jobs in decreasing order of their runtime. 3. Create a heap containing clusters.num number of clustered jobs. 4. For each job j, repeat a. Get cluster job cj, having the shortest runtime b. Add job j to clustered job cj
The runtime of a job, and the number of clustered jobs to create is determined by the value of two parameters associated with the jobs.
runtime
expected runtime for a job
clusters.num
clusters.numfactor denotes how many clustered jobs does the user want to see per level per site
Note
Users should either specify clusters.maxruntime or clusters.num. If
both of them are specified, then clusters.num profile will be ignored
by the clustering engine.
All of these parameters are specified by the use of a PEGASUS namespace
profile key. The keys can be specified at any of the placeholders for
the profiles (abstract transformation in the Abstract Workflow, site in the site
catalog, transformation in the transformation catalog). The normal
overloading semantics apply i.e. profile in transformation catalog
overrides the one in the site catalog and that in turn overrides the one
in the Abstract Workflow. The two parameters are described below.
#!/usr/bin/env python3 from Pegasus.api import * # create the TransformationCatalog object tc = TransformationCatalog() # create and add the transformation # Cluster all jobs of type B at siteX, into 2 clusters # such that the 2 clusters have similar runtimes B = Transformation( "B", site="siteX", pfn="/shared/PEGASUS/bin/jobB", is_stageable=False, ).add_profiles(Namespace.PEGASUS, key="clusters.num", value=2)\ .add_profiles(Namespace.PEGASUS, key="runtime", value=100) tc.add_transformations(B) # Cluster all jobs of type C at siteX, such that the duration # duration of the clustered job does not exceed 300. C = Transformation( "C", site="siteX", pfn="/shared/PEGASUS/bin/jobC", is_stageable=False, ).add_profiles(Namespace.PEGASUS, key="maxruntime", value=300)\ .add_profiles(Namespace.PEGASUS, key="runtime", value=100) tc.add_transformations(C) # write the transformation catalog to the default file path "./transformations.yml" tc.write()x-pegasus: {apiLang: python, createdBy: vahi, createdOn: '10-29-20T14:45:49Z'} pegasus: '5.0' transformations: - name: B sites: - {name: siteX, pfn: /shared/PEGASUS/bin/jobB, type: installed} profiles: pegasus: {clusters.num: 2, runtime: 100} - name: C sites: - {name: siteX, pfn: /shared/PEGASUS/bin/jobC, type: installed} profiles: pegasus: {maxruntime: 300, runtime: 100}# multiple line text-based transformation catalog: 2014-09-30T16:09:40.610-07:00 #Cluster all jobs of type B at siteX, into 2 clusters such that the 2 clusters have similar runtimes tr B { site siteX { profile pegasus "clusters.num" "2" profile pegasus "runtime" "100" pfn "/shared/PEGASUS/bin/jobB" arch "x86" os "LINUX" type "INSTALLED" } } #Cluster all jobs of type C at siteX, such that the duration of the clustered job does not exceed 300. tr C { site siteX { profile pegasus "clusters.maxruntime" "300" profile pegasus "runtime" "100" pfn "/shared/PEGASUS/bin/jobC" arch "x86" os "LINUX" type "INSTALLED" } }
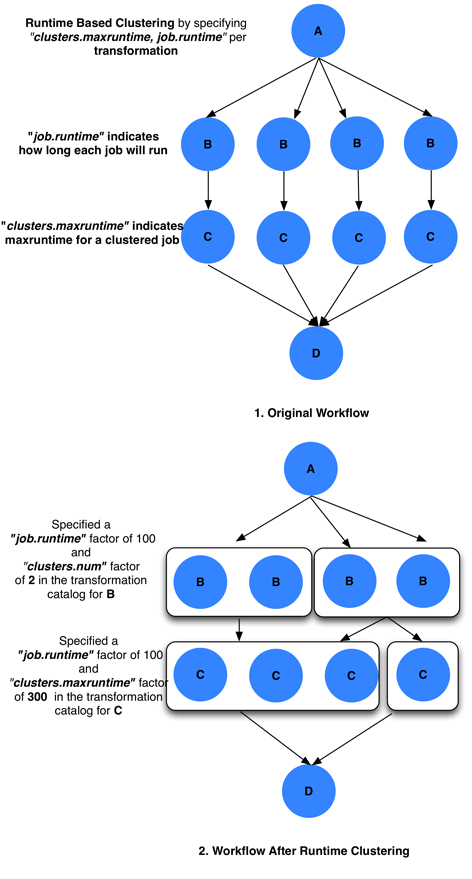
Clustering by runtime
In the above case the jobs referring to logical transformation B
scheduled on siteX will be clustered such that all clustered jobs will
run approximately for the same duration specified by the
clusters.maxruntime property. In the above case we assume all jobs
referring to transformation B run for 100 seconds. For jobs with
significantly differing runtime, the runtime property will be associated
with the jobs in the Abstract Workflow.
In addition to the above two profiles, we need to inform pegasus-plan to use runtime clustering. This is done by setting the following property .
pegasus.clusterer.preference Runtime
12.2.5. Label Clustering
In label based clustering, the user labels the workflow. All jobs having the same label value are clustered into a single clustered job. This allows the user to create clusters or use a clustering technique that is specific to his or her workflow. If there is no label associated with the job, the job is not clustered and is executed as is
Since, the jobs in a cluster in this case are not independent, it is
important the jobs are executed in the correct order. This is done by
doing a topological sort on the jobs in each cluster. To use label based
clustering the user needs to set the --cluster option of
pegasus-plan to label.
12.2.5.1. Labelling the Workflow
The labels for the jobs in the workflow are specified by associated
PEGASUS profile keys with the jobs during the Abstract Workflow generation
process. The user can choose which profile key to use for labeling the
workflow. By default, it is assumed that the user is using the PEGASUS
profile key label to associate the labels. To use another key, in the
PEGASUS namespace the user needs to set the following property
pegasus.clusterer.label.key
For example if the user sets pegasus.clusterer.label.key to
user_label then the job description in the Abstract Workflow looks as follows
<adag >
...
<job id="ID000004" namespace="app" name="analyze" version="1.0" level="1" >
<argument>-a bottom -T60 -i <filename file="user.f.c1"/> -o <filename file="user.f.d"/></argument>
<profile namespace="pegasus" key="user_label">p1</profile>
<uses file="user.f.c1" link="input" register="true" transfer="true"/>
<uses file="user.f.c2" link="input" register="true" transfer="true"/>
<uses file="user.f.d" link="output" register="true" transfer="true"/>
</job>
...
</adag>
The above states that the
PEGASUSprofiles with key asuser_labelare to be used for designating clusters.Each job with the same value for
PEGASUSprofile keyuser_labelappears in the same cluster.
12.2.6. Whole Clustering
In whole workflow clustering, all the jobs in the workflow get clustered into
a single job. This clustering is a specialized case of label based clustering
where all jobs in the workflow are assumed to have the same label. This is
particularly useful when you want to run the whole workflow using PMC.
To use whole workflow clustering the user needs to set the --cluster option
of pegasus-plan to whole.
12.2.7. Recursive Clustering
In some cases, a user may want to use a combination of clustering
techniques. For e.g. a user may want some jobs in the workflow to be
horizontally clustered and some to be label clustered. This can be
achieved by specifying a comma separated list of clustering techniques
to the --cluster option of pegasus-plan. In this case the
clustering techniques are applied one after the other on the workflow in
the order specified on the command line.
For example
$ pegasus-plan --dir ./dags --cluster label,horizontal -s siteX --output local --verbose workflow.yml
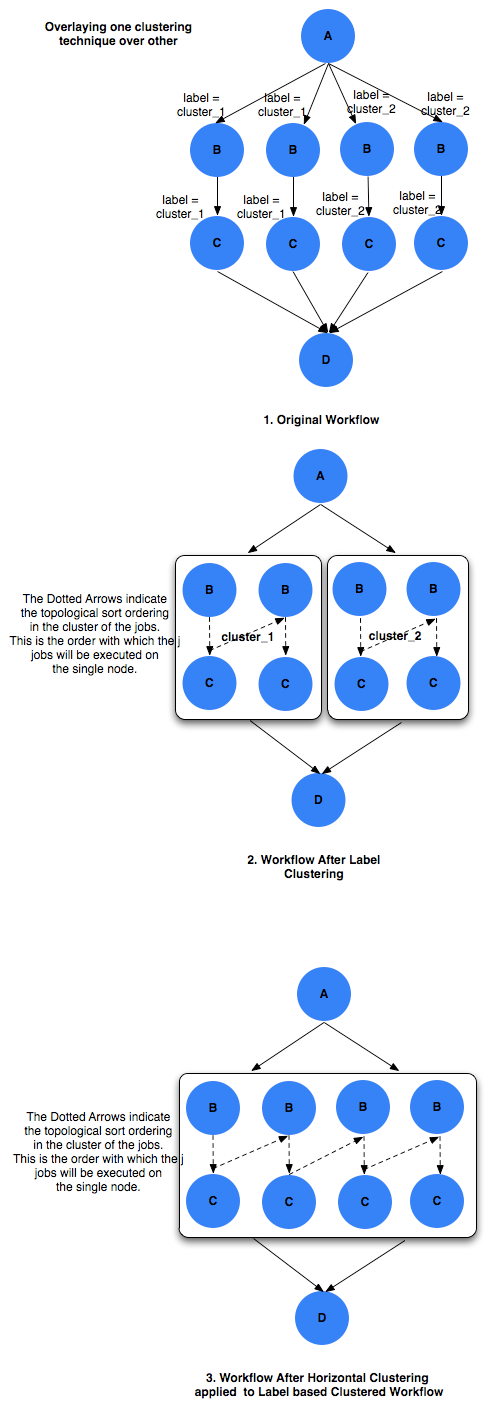
Recursive clustering
12.2.8. Execution of the Clustered Job
The execution of the clustered job on the remote site, involves the execution of the smaller constituent jobs either
sequentially on a single node of the remote site
The clustered job is executed using pegasus-cluster, a wrapper tool written in C that is distributed as part of Pegasus. It takes in the jobs passed to it, and ends up executing them sequentially on a single node. To use pegasus-cluster for executing any clustered job on a siteX, there needs to be an entry in the transformation catalog for an executable with the logical name seqexec and namespace as pegasus.
#!/usr/bin/env python3 from Pegasus.api import * # create the TransformationCatalog object tc = TransformationCatalog() seqexec = Transformation( "pegasus", "seqexec", site="siteX", pfn="/user/bin/pegasus-cluster", arch=Arch.X86_64, is_stageable=False ) tc.add_transformations(seqexec) # write the transformation catalog to the default file path "./transformations.yml" tc.write()
x-pegasus: {apiLang: python, createdBy: vahi, createdOn: '10-29-20T14:57:48Z'} pegasus: '5.0' transformations: - namespace: seqexec name: pegasus sites: - {name: siteX, pfn: /user/bin/pegasus-cluster, type: installed, arch: x86_64}
tr pegasus::seqexec { site siteX { pfn "/user/bin/pegasus-cluster" arch "x86_64" os "LINUX" type "INSTALLED" } }
If the entry is not specified, Pegasus will attempt create a default path on the basis of the environment profile
PEGASUS_HOMEspecified in the site catalog for the remote site.On multiple nodes of the remote site using MPI based task management tool called Pegasus MPI Cluster (PMC)
The clustered job is executed using pegasus-mpi-cluster, a wrapper MPI program written in C that is distributed as part of Pegasus. A PMC job consists of a single master process (this process is rank 0 in MPI parlance) and several worker processes. These processes follow the standard master-worker architecture. The master process manages the workflow and assigns workflow tasks to workers for execution. The workers execute the tasks and return the results to the master. Communication between the master and the workers is accomplished using a simple text-based protocol implemented using MPI_Send and MPI_Recv. PMC relies on a shared filesystem on the remote site to manage the individual tasks stdout and stderr and stage it back to the submit host as part of it’s own stdout/stderr.
The input format for PMC is a DAG based format similar to Condor DAGMan’s. PMC follows the dependencies specified in the DAG to release the jobs in the right order and executes parallel jobs via the workers when possible. The input file for PMC is automatically generated by the Pegasus Planner when generating the executable workflow. PMC allows for a finer grained control on how each task is executed. This can be enabled by associating the following pegasus profiles with the jobs in the Abstract Workflow
Pegasus Profiles that can be associated with jobs in the Abstract Workflow for PMC Key
Description
pmc_request_memory
This key is used to set the -m option for pegasus-mpi-cluster. It specifies the amount of memory in MB that a job requires. This profile is usually set in the Abstract Workflow for each job.
pmc_request_cpus
This key is used to set the -c option for pegasus-mpi-cluster. It specifies the number of cpu’s that a job requires. This profile is usually set in the Abstract Workflow for each job.
pmc_priority
This key is used to set the -p option for pegasus-mpi-cluster. It specifies the priority for a job . This profile is usually set in the Abstract Workflow for each job. Negative values are allowed for priorities.
pmc_task_arguments
The key is used to pass any extra arguments to the PMC task during the planning time. They are added to the very end of the argument string constructed for the task in the PMC file. Hence, allows for overriding of any argument constructed by the planner for any particular task in the PMC job.
Refer to the pegasus-mpi-cluster manpage to learn more about how it schedules individual tasks.
It is recommended to have a
pegasus::mpiexecentry in the transformation catalog to specify the path to PMC on the remote and specify the relevant globus profiles such asxcount,host_xcountandmaxwalltimeto control size of the MPI job.#!/usr/bin/env python3 from Pegasus.api import * # create the TransformationCatalog object tc = TransformationCatalog() pmc = Transformation( "pegasus", "mpiexec", site="siteX", pfn="/usr/bin/pegasus-mpi-cluster", is_stageable=False, ).add_profiles(Namespace.PEGASUS, key="nodes", value=1)\ .add_profiles(Namespace.PEGASUS, key="ppn", value=32) tc.add_transformations(pmc) # write the transformation catalog to the default file path "./transformations.yml" tc.write()
x-pegasus: {apiLang: python, createdBy: vahi, createdOn: '10-29-20T14:57:48Z'} pegasus: '5.0' transformations: - namespace: mpiexec name: pegasus sites: - {name: siteX, pfn: /usr/bin/pegasus-mpi-cluster, type: installed} profiles: pegasus: {nodes: 1, ppn: 32}
tr pegasus::mpiexec { site siteX { pfn "/user/bin/pegasus-mpi-cluster" arch "x86_64" os "LINUX" type "INSTALLED" profile pegasus nodes 1 profile pegasus ppn 32 } }
If the entry is not specified, Pegasus will attempt create a default path on the basis of the environment profile
PEGASUS_HOMEspecified in the site catalog for the remote site.Tip
Users are encouraged to use label based clustering in conjunction with PMC
On multiple nodes of the remote site using MPI based in-situ task management tool called Decaf
Decaf is a middleware for building and executing in-situ workflows. Decaf allows parallel communication of coupled tasks by creating communication channels over HPC interconnects through MPI. Decaf has a Python API, where users can describe the workflow graph by defining the tasks and the communication channels among them. Decaf does not impose any constraints on this graph topology and can manage graphs with cycles. Once the workflow graph is defined, it is executed as a multiple-program-multiple-data (MPMD) MPI application.
To use in-situ frameworks such as Decaf the underlying application code needs to be changed to use the Decaf libraries and constructs for their file I/O. This cannot be avoided. However, Pegasus ensures that users don’t have to change their workflow generators to use Decaf. Decaf is integrated into Pegasus as a technique to manage execution of clustered jobs. When users enable job clustering in their workflows at planning time, Pegasus can set up a sub graph of the workflow (identified using label based clustering) to be clustered and executed using Decaf. As part of this process, Pegasus creates the necessary bindings for the clustered job to execute using Decaf.
To execute a clustered job using Decaf, Pegasus generates
the JSON file for the workflow graph, and
the run script for the users to run the Decaf via mpmd.
To automatically generate the Decaf description Pegasus employs the following rules
the roots of the sub graph will have no inports and the leaves of the sub graph have no outports
the func name is generated based on the transformation name the node maps to for link jobs
the name attribute is derived from the func attribute of the nodes making up the edges
the source and target are the decaf integer id’s of the nodes.
also internally order for the jobs is preserved as specified in the input workflow description.
In addition, Pegasus generates a shell script for the clustered job that allows the clustered DECAF job to be run via SLURM. Below is an actual script from one of the runs of 1000Genome workflow that was executed on CORI.
#!/bin/bash set -e LAUNCH_DIR=`pwd` echo "Job Launched in directory $LAUNCH_DIR" source $DECAF_ENV_SOURCE # copy the json file for the job into the directory # where we are going to launch decaf cp 1Kgenome.json $PEGASUS_SCRATCH_DIR/ cd $PEGASUS_SCRATCH_DIR echo "Invoking decaf executable from directory `pwd`" cat <<EOF > merge_cluster1.conf 0 ./individuals ALL.chr1.250000.vcf 1 1 15626 250000 1 ./individuals ALL.chr1.250000.vcf 1 171876 187501 250000 2 ./individuals ALL.chr1.250000.vcf 1 15626 31251 250000 3 ./individuals ALL.chr1.250000.vcf 1 187501 203126 250000 4 ./individuals ALL.chr1.250000.vcf 1 140626 156251 250000 5 ./individuals ALL.chr1.250000.vcf 1 156251 171876 250000 6 ./individuals ALL.chr1.250000.vcf 1 62501 78126 250000 7 ./individuals ALL.chr1.250000.vcf 1 234376 250001 250000 8 ./individuals ALL.chr1.250000.vcf 1 78126 93751 250000 9 ./individuals ALL.chr1.250000.vcf 1 31251 46876 250000 10 ./individuals ALL.chr1.250000.vcf 1 203126 218751 250000 11 ./individuals ALL.chr1.250000.vcf 1 46876 62501 250000 12 ./individuals ALL.chr1.250000.vcf 1 218751 234376 250000 13 ./individuals ALL.chr1.250000.vcf 1 125001 140626 250000 14 ./individuals ALL.chr1.250000.vcf 1 93751 109376 250000 15 ./individuals ALL.chr1.250000.vcf 1 109376 125001 250000 16 ./individuals_merge 1 chr1n-1-15626.tar.gz chr1n-15626-31251.tar.gz chr1n-31251-46876.tar.gz chr1n-46876-62501.tar.gz chr1n-62501-78126.tar.gz chr1n-78126-93751.tar.gz chr1n-93751-109376.tar.gz chr1n-109376-125001.tar.gz chr1n-125001-140626.tar.gz chr1n-140626-156251.tar.gz chr1n-156251-171876.tar.gz chr1n-171876-187501.tar.gz chr1n-187501-203126.tar.gz chr1n-203126-218751.tar.gz chr1n-218751-234376.tar.gz chr1n-234376-250001.tar.gz EOF srun --multi-prog ./merge_cluster1.conf
You need to have a
dataflow::decafentry in the transformation catalog to specify basename of the json file that you want created for execution on the remote site#!/usr/bin/env python3 from Pegasus.api import * # create the TransformationCatalog object tc = TransformationCatalog() n_nodes = 17 decaf = Transformation("decaf", namespace="dataflow", site="cori", pfn=json_fn, is_stageable=False) .add_pegasus_profile( runtime="18000", glite_arguments="--qos=regular --constraint=haswell --licenses=SCRATCH --nodes=" + str(n_nodes) + " --ntasks-per-node=1 --ntasks=" + str(n_nodes), # glite_arguments="--qos=debug --constraint=haswell --licenses=SCRATCH", # exitcode.successmsg="Execution time in seconds:", ) .add_profiles(Namespace.PEGASUS, key="exitcode.successmsg", value="Execution time in seconds:") .add_profiles(Namespace.PEGASUS, key="dagman.post", value="pegasus-exitcode") .add_env(key="DECAF_ENV_SOURCE", value=env_script) tc.add_transformations(decaf) # write the transformation catalog to the default file path "./transformations.yml" tc.write()
x-pegasus: apiLang: python createdBy: pegasus createdOn: 10-17-21T15:23:28Z pegasus: '5.0' transformations: - namespace: dataflow name: decaf sites: - name: cori pfn: 1Kgenome.json type: installed profiles: pegasus: runtime: '12000' glite.arguments: --qos=regular --constraint=haswell --licenses=SCRATCH --nodes=17 --ntasks-per-node=1 --ntasks=17 exitcode.successmsg: 'Execution time in seconds:' dagman.post: pegasus-exitcode env: DECAF_ENV_SOURCE: /global/cfs/cdirs/m2187/pegasus-decaf/1000genome-workflow/env.sh
Note
If you want to use Decaf for your workflows, please contact the Decaf team at Argonne for help on how to port your application to Decaf.
12.2.9. Specification of Method of Execution for Clustered Jobs
The method execution of the clustered job(whether to launch via mpiexec or seqexec) can be specified
globally in the properties file
The user can set a property in the properties file that results in all the clustered jobs of the workflow being executed by the same type of executable.
#PEGASUS PROPERTIES FILE pegasus.clusterer.job.aggregator seqexec|mpiexec
In the above example, all the clustered jobs on the remote sites are going to be launched via the property value, as long as the property value is not overridden in the site catalog.
associating profile key job.aggregator with the site in the site catalog
<site handle="siteX" gridlaunch = "/shared/PEGASUS/bin/kickstart"> <profile namespace="env" key="GLOBUS_LOCATION" >/home/shared/globus</profile> <profile namespace="env" key="LD_LIBRARY_PATH">/home/shared/globus/lib</profile> <profile namespace="pegasus" key="job.aggregator" >seqexec</profile> <lrc url="rls://siteX.edu" /> <gridftp url="gsiftp://siteX.edu/" storage="/home/shared/work" major="2" minor="4" patch="0" /> <jobmanager universe="transfer" url="siteX.edu/jobmanager-fork" major="2" minor="4" patch="0" /> <jobmanager universe="vanilla" url="siteX.edu/jobmanager-condor" major="2" minor="4" patch="0" /> <workdirectory >/home/shared/storage</workdirectory> </site>
In the above example, all the clustered jobs on a siteX are going to be executed via seqexec, as long as the value is not overridden in the transformation catalog.
associating profile key job.aggregator with the transformation that is being clustered, in the transformation catalog
#!/usr/bin/env python3 from Pegasus.api import * # create the TransformationCatalog object tc = TransformationCatalog() # create and add the transformation B = Transformation( "B", site="siteX", pfn="/shared/PEGASUS/bin/jobB", is_stageable=False, ).add_profiles(Namespace.PEGASUS, key="clusters.num", value=2)\ .add_profiles(Namespace.PEGASUS, key="job.aggregator", value="mpiexec") tc.add_transformations(B) # write the transformation catalog to the default file path "./transformations.yml" tc.write()
x-pegasus: {apiLang: python, createdBy: vahi, createdOn: '10-29-20T14:45:49Z'} pegasus: '5.0' transformations: - name: B sites: - {name: siteX, pfn: /shared/PEGASUS/bin/jobB, type: installed} profiles: pegasus: {clusters.num: 2, job.aggregator: "mpiexec"}
# multiple line text-based transformation catalog: 2014-09-30T16:09:40.610-07:00 # jobs of type B when clustered, should run using pegasus-mpi-cluster tr B { site siteX { profile pegasus "clusters.num" "2" profile pegasus "job.aggregator" "mpiexec" pfn "/shared/PEGASUS/bin/jobB" arch "x86" os "LINUX" type "INSTALLED" } }
In the above example, all the clustered jobs that consist of transformation B on siteX will be executed via mpiexec.
Note
The clustering of jobs on a site only happens only if
there exists an entry in the transformation catalog for the clustering executable that has been determined by the above 3 rules
the number of jobs being clustered on the site are more than 1
12.2.10. Outstanding Issues
Label Clustering
More rigorous checks are required to ensure that the labeling scheme applied by the user is valid.
12.3. Hierarchical Workflows
Issue: When planning and running large workflows, there are some scalability issues to be aware of. During the planning stage, Pegasus traverses the graphs multiple times, and some of the graph transforms can be slow depending on how large the graph is regarding the number of tasks, files, and dependencies. Once planned, large workflows can also see scalability limits when interacting with the operating system. A common problem is the number of files in a single directory, such as thousands or millons of input or output files.
Solution: The most common solution to these problems is to use hierarchical workflows, which works really well if your workflow can be logically partitioned into smaller workflows. A hierarchical workflow still runs like a single workflow, with the difference being that some jobs in the workflow are actually sub-workflows.
The Abstract Workflow in addition to containing compute jobs, can also contain jobs that refer to other workflows. This is useful for running large workflows or ensembles of workflows.
Users can embed two types of workflow jobs in the Abstract Workflow
pegasusWorkflow - refers to a sub workflow represented as an Abstract Workflow (one which is generated by one of the provided APIs). During the planning of a workflow, the pegasusWorkflow jobs are mapped to condor dagman jobs that have a pegasus-plan invocation on the Abstract Workflow for that sub workflow set as the prescript.
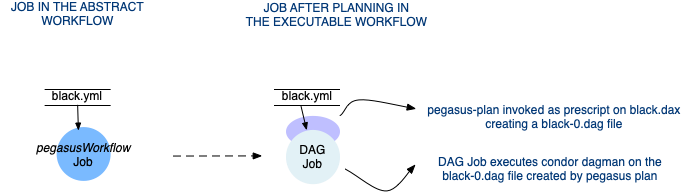
Planning of a pegasusWorkflow Job
condorWorkflow - refers to a sub workflow represented as a DAG. During the planning of a workflow, the DAG jobs are mapped to condor dagman and refer to the DAG file mentioned in the DAG job.

Planning of a condorWorkflow Job
12.3.1. Specifying a pegasusWorkflow Job in the Abstract Workflow
Specifying a pegasusWorkflow in a Abstract Workflow is pretty similar to
how normal compute jobs are specified. There are minor differences in
terms of the yaml element name ( pegasusWorkflow vs job ) and the
attributes specified.
An example pegasusWorkflow Job in the Abstract Workflow is shown below
#!/usr/bin/env python3
from Pegasus.api import *
wf = Workflow("local-hierarchy")
fd = File("f.d")
blackdiamond_wf = SubWorkflow("blackdiamond.yml", False).add_args(
"--input-dir", "input", "--output-sites", "local", "-vvv", "--force"
).add_outputs(fd).add_dagman_profile(max_jobs="10")
wf.add_jobs(blackdiamond_wf)
# writes out to workflow.yml
wf.write()
x-pegasus: {apiLang: python, createdBy: vahi, createdOn: '10-29-20T16:42:51Z'}
pegasus: '5.0'
name: local-hierarchy
jobs:
- type: pegasusWorkflow
file: blackdiamond.yml
id: ID0000001
arguments: [--input-dir, input, --output-sites, local, -vvv, --force]
uses:
- {lfn: blackdiamond.yml, type: input}
- {lfn: f.d, type: output, stageOut: true, registerReplica: true}
profiles:
dagman: {MAXJOBS: '10'}
jobDependencies: []
12.3.1.1. Abstract Workflow File Locations
The file key for the pegasusWorkflow job refers to the LFN ( Logical File Name ) of the Abstract Workflow file. The location of the Abstract Workflow file can be catalogued either in the
Replica Catalog
Replica Catalog Section in the Abstract Workflow.
Note
Currently, only file url’s on the local site ( submit host ) can be specified as Abstract Workflow File locations.
12.3.1.2. Generating Abstract Workflow as a Compute Job
It is possible, to have the abstract workflow that a pegasusWorkflow job requires, to be a parent compute job in the hierarchical workflow. This is useful, when the generation of the abstract workflow the pegasusWorkflow
takes a lot of resources (time/memory) to generate.
the abstract workflows cannot be pre-generated when defining the hierarchical workflow.
In these scenarios, you can have a compute job generate the abstract workflow for the sub-workflows. The code snippets highlight it below
#!/usr/bin/env python3
from Pegasus.api import *
wf = Workflow("hierarchical-workflow")
# job to generate the diamond workflow
diamond_wf_file = File("inner_diamond_workflow.yml")
generate_diamond_wf_job = Job(
generate_diamond_wf, _id="diamond_workflow_gen"
).add_outputs(diamond_wf_file)
# job to plan and run the diamond workflow
# the file diamond_wf_file is set to be create by the
# generate_diamond_wf_job
diamond_wf_job = (
SubWorkflow(file=diamond_wf_file, is_planned=False, _id="diamond_subworkflow")
.add_args(
"--conf",
"inner_diamond_workflow.pegasus.properties",
"--output-sites",
"local",
"-vvv",
"--basename",
"inner",
)
.add_inputs(
File("inner_diamond_workflow.pegasus.properties", for_planning=True),
File("inner_diamond_workflow_rc.yml", for_planning=True),
File("inner_diamond_workflow_tc.yml", for_planning=True),
File("sites.yml", for_planning=True),
)
)
wf.add_jobs(blackdiamond_wf)
# writes out to workflow.yml
wf.write()
x-pegasus: {apiLang: python, createdBy: bamboo, createdOn: '03-07-23T14:24:45Z'}
pegasus: 5.0.4
name: hierarchical-workflow
jobs:
- type: job
name: generate_inner_diamond_workflow.py
id: diamond_workflow_gen
arguments: []
uses:
- {lfn: inner_diamond_workflow.yml, type: output, stageOut: true, registerReplica: true}
- type: pegasusWorkflow
file: inner_diamond_workflow.yml
id: diamond_subworkflow
arguments: [--conf, inner_diamond_workflow.pegasus.properties, --output-sites, local,
-vvv, --basename, inner]
uses:
- {lfn: inner_diamond_workflow_rc.yml, forPlanning: true, type: input}
- {lfn: inner_diamond_workflow.yml, forPlanning: true, type: input}
- {lfn: inner_diamond_workflow.pegasus.properties, forPlanning: true, type: input}
- {lfn: inner_diamond_workflow_tc.yml, forPlanning: true, type: input}
- {lfn: sites.yml, forPlanning: true, type: input}
jobDependencies:
- id: diamond_workflow_gen
children: [diamond_subworkflow]
12.3.1.3. Arguments for a pegasusWorkflow Job
Users can specify specific arguments for pegasusWorkflow jobs. The arguments specified for the pegasusWorkflow jobs are passed to the pegasus-plan invocation in the prescript for the corresponding condor dagman job in the executable workflow.
The following options for pegasus-plan are inherited from the pegasus-plan invocation of the parent workflow. If an option is specified in the arguments section for the pegasusWorkflow job then that overrides what is inherited.
Option Name |
Description |
|---|---|
–sites |
list of execution sites. |
It is highly recommended that users don’t specify directory related options in the arguments section for the DAX Jobs. Pegasus assigns values to these options for the sub workflows automatically.
--relative-dir--dir--relative-submit-dir
12.3.1.4. Profiles for pegasusWorkflow Job
Users can choose to specify dagman profiles with the pegasusWorkflow job to
control the behavior of the corresponding condor dagman instance in the
executable workflow. In the example above
maxjobs is set to 10 for the sub workflow.
12.3.1.5. Catalogs in Hierarchical Workflows
When using hierarchical workflows, and you want to use the same catalog files for all the workflows making up your hierarchical workflow it is advisable to have the catalog files as standalone catalog files, and locations of those catalogued in your properties.
Catalogs defined inline in the abstract workflow are not inherited by a sub-workflow. The only exception to this is the replica catalog that is inherited one level ( if in a worklfow W you have a replica catalog inlined, the worklfows corresponding to the pegasusWorkflow jobs defined in W will have access to replica catalog defined in W).
12.3.1.6. Execution of the PRE script and HTCondor DAGMan instance
The pegasus-plan that is invoked as part of the prescript to the condor dagman job is executed on the submit host. The log from the output of pegasus-plan is redirected to a file ( ending with suffix pre.log ) in the submit directory of the workflow that contains the DAX Job. The path to pegasus-plan is automatically determined.
The pegasusWorkflow job maps to a Condor DAGMan job. The path to condor dagman binary is determined according to the following rules -
entry in the transformation catalog for condor::dagman for site local, else
pick up the value of
CONDOR_HOMEfrom the environment if specified and set path to condor dagman as$CONDOR_HOME/bin/condor_dagman, elsepick up the value of
CONDOR_LOCATIONfrom the environment if specified and set path to condor dagman as$CONDOR_LOCATION/bin/condor_dagman, elsepick up the path to condor dagman from what is defined in the user’s
PATH
Tip
It is recommended that users specify dagman.maxpre in their
properties file to control the maximum number of pegasus-plan
instances launched by each running dagman instance.
12.3.2. Specifying a condorWorkflow Job in the Abstract Workflow
Specifying a condorWorkflow in an Abstract Workflow is pretty similar to how
normal compute jobs are specified. There are minor differences in terms
of the yaml element name ( condorWorkflow vs job ) and the attributes
specified. An example condorWorkflow
job in an Abstract Workflow is shown below
#!/usr/bin/env python3
from Pegasus.api import *
wf = Workflow("local-hierarchy")
blackdiamond_wf = SubWorkflow("black.dag", True).add_dagman_profile(max_jobs="10").add_profiles(
Namespace.DAGMAN, key="dir", value="/dag-dir/test")
wf.add_jobs(blackdiamond_wf)
# writes out to workflow.yml
wf.write()
x-pegasus: {apiLang: python, createdBy: vahi, createdOn: '10-29-20T17:10:43Z'}
pegasus: '5.0'
name: local-hierarchy
jobs:
- type: condorWorkflow
file: black.dag
id: ID0000001
arguments: []
uses:
- {lfn: black.dag, type: input}
profiles:
dagman: {MAXJOBS: '10', dir: /dag-dir/test}
jobDependencies: []
12.3.2.1. DAG File Locations
The name attribute in the condorWorkflow element refers to the LFN ( Logical File Name ) of the HTCondor dag file. The location of the DAG file can be catalogued either in the
Replica Catalog
Replica Catalog Section in the Abstract Workflow.
Note
Currently, only file url’s on the local site ( submit host ) can be specified as DAG file locations.
12.3.2.2. Profiles for condorWorkflow Job
Users can choose to specify dagman profiles with the condorWorkflow job
to control the behavior of the corresponding condor dagman instance in the
executable workflow. In the example above, maxjobs is set to 10 for the
sub workflow.
The dagman profile DIR allows users to specify the directory in which
they want the condor dagman instance to execute. In the example
above black.dag is set to be executed in
directory /dag-dir/test . The /dag-dir/test should be created
beforehand.
12.3.3. File Dependencies in Hierarchical Workflows
In this section, we explain file dependencies in hierarchical workflows namely
data dependencies between pegasusWorkflow jobs
data dependency between a pegasusWorkflow job and a compute job
Note
Starting with Pegasus 5.x releases, it is recommended that you list the file dependencies for a pegasusWorkflow job as you would do with a normal compute job. Additionally, the sub workflows no longer get passed the cache file generated when the planning the enclosing workflow ( the workflow in which you define the pegasusWorkflow jobs). Hence it is advisable to list any file dependencies that a pegasusWorkflow job may have to a compute job or other pegasusWorkflow jobs.
12.3.3.1. File Dependencies Across pegasusWorkflow Jobs
In hierarchical workflows , if a sub workflow generates some output files required by another sub workflow then there should be an edge connecting the two pegasusWorkflow jobs. Pegasus will ensure that the prescript for the child sub-workflow, has the path to the cache file generated during the planning of the parent sub workflow. The cache file in the submit directory for a workflow is a textual replica catalog that lists the locations of all the output files created in the remote workflow execution directory when the workflow executes.
This automatic passing of the cache file to a child sub-workflow ensures that the datasets from the same workflow run are used. However, the passing the locations in a cache file also ensures that Pegasus will prefer them over all other locations in the Replica Catalog. If you need the Replica Selection to consider locations in the Replica Catalog also, then set the following property.
pegasus.catalog.replica.cache.asrc true
The above is useful in the case, where you are staging out the output files to a storage site, and you want the child sub workflow to stage these files from the storage output site instead of the workflow execution directory where the files were originally created.
#!/usr/bin/env python3
from Pegasus.api import *
# define various transformations.
...
# --- SubWorkflow1 ---------------------------------------------------------------
input_file = File("input.txt")
k1_out = File("k1.txt")
wf1 = Workflow("subworkflow-1")
k1 = Job(keg)\
.add_args("-i", input_file, "-o", k1_out, "-T", 5)\
.add_inputs(input_file)\
.add_outputs(k1_out)
ls1 = Job(ls)\
.add_args("-alh")
wf1.add_jobs(k1, ls1)
wf1.write("subwf1.yml")
# --- SubWorkflow2 ---------------------------------------------------------------
k2_out = File("k2.txt")
wf2 = Workflow("subworkflow-2")
k2 = Job(keg)\
.add_args("-i", k1_out, "-o", k2_out, "-T", 5)\
.add_inputs(k1_out)\
.add_outputs(k2_out)
wf2.add_jobs(k2)
wf2.write("subwf2.yml")
# Root
root_wf = Workflow("root")
j1 = SubWorkflow("subwf1.yml", _id="subwf1")\
.add_planner_args(verbose=3)\
.add_outputs(k1_out)
j2 = SubWorkflow("subwf2.yml", _id="subwf2")\
.add_planner_args(verbose=3)\
.add_inputs(k1_out)\
.add_outputs(k2_out)
root_wf.add_jobs(j1, j2)
x-pegasus: {apiLang: python, createdBy: bamboo, createdOn: '10-05-21T09:27:22Z'}
pegasus: '5.0'
name: root
jobs:
- type: pegasusWorkflow
file: subwf1.yml
id: subwf1
arguments: [-vvv]
uses:
- {lfn: subwf1.yml, type: input}
- {lfn: k1.txt, type: output, stageOut: true, registerReplica: true}
- type: pegasusWorkflow
file: subwf2.yml
id: subwf2
arguments: [-vvv]
uses:
- {lfn: k2.txt, type: output, stageOut: true, registerReplica: true}
- {lfn: subwf2.yml, type: input}
- {lfn: k1.txt, type: input}
jobDependencies:
- id: subwf1
children: [subwf2]
12.3.3.2. File Dependencies between pegasusWorkflow and Compute Jobs
If in the same workflow W, you have a pegasusWorkflow job (Job D) and a dependant compute job (Job C), where job C requires as input an output file that is created when the sub workflow corresponding to Job D is run; then the planner will ensure that when the sub workflow corresponding to Job D runs, it also transfers that file to the scratch directory on the staging site for workflow W, in addition to transferring to the output site.
This is achieved by passing an output.map file to the pegasusWorkflow job that lists the location on the staging site (where job D will pickup from when it executes). The output.map file tells the pegasusWorkflow job as to where to place certain outputs.
#!/usr/bin/env python3
from Pegasus.api import *
fa = File("f.a")
fb1 = File("f.b1")
fb2 = File("f.b2")
fc1 = File("f.c1")
fc2 = File("f.c2")
fd = File("f.d")
fe = File("f.e")
# define various transformations.
...
# --- Blackdiamond Sub Workflow -------------------------------------------------------
wf = (
Workflow("blackdiamond")
.add_jobs(
Job("preprocess", namespace="diamond", version="4.0")
.add_args("-a", "preprocess", "-T", "60", "-i", fa, "-o", fb1, fb2)
.add_inputs(fa)
.add_outputs(fb1, fb2, register_replica=True),
Job("findrange", namespace="diamond", version="4.0")
.add_args("-a", "findrange", "-T", "60", "-i", fb1, "-o", fc1)
.add_inputs(fb1)
.add_outputs(fc1, register_replica=True),
Job("findrange", namespace="diamond", version="4.0")
.add_args("-a", "findrange", "-T", "60", "-i", fb2, "-o", fc2)
.add_inputs(fb2)
.add_outputs(fc2, register_replica=True),
Job("analyze", namespace="diamond", version="4.0")
.add_args("-a", "analyze", "-T", "60", "-i", fc1, fc2, "-o", fd)
.add_inputs(fc1, fc2)
.add_outputs(fd, register_replica=False, stage_out=False),
)
.write(str(TOP_DIR / "input/blackdiamond.yml"))
)
# --- Top Root Level Workflow -------------------------------------------------------
wf = Workflow("local-hierarchy")
blackdiamond_wf = SubWorkflow("blackdiamond.yml", False).add_args(
"--input-dir", "input", "--output-sites", "local", "-vvv", "--force"
).add_outputs(fd)
# --- Compute Job that is dependent on output f.d created by the Sub Workflow-------
post_analyze_job = Job("post-analyze", namespace="diamond", version="4.0")\
.add_args("-a", "post-analyze", "-T", "60", "-i", fd, "-o", fe)\
.add_inputs(fd)\
.add_outputs(fe, register_replica=True, stage_out=True)
wf.add_jobs(blackdiamond_wf, post_analyze_job)
wf.add_dependency(blackdiamond_wf, children=[post_analyze_job])
x-pegasus: {apiLang: python, createdBy: bamboo, createdOn: '10-05-21T09:27:21Z'}
pegasus: '5.0'
name: local-hierarchy
jobs:
- type: pegasusWorkflow
file: blackdiamond.yml
id: ID0000001
arguments: [--input-dir, input, --output-sites, local, -vvv, --force]
uses:
- {lfn: f.d, type: output, stageOut: true, registerReplica: true}
- {lfn: blackdiamond.yml, type: input}
- type: job
namespace: diamond
version: '4.0'
name: post-analyze
id: ID0000002
arguments: [-a, post-analyze, -T, '60', -i, f.d, -o, f.e]
uses:
- {lfn: f.d, type: input}
- {lfn: f.e, type: output, stageOut: true, registerReplica: true}
jobDependencies:
- id: ID0000001
children: [ID0000002]
12.3.4. Recursion in Hierarchical Workflows
It is possible for a user to add a jobs to a workflow that already contains jobs in them. Pegasus does not place a limit on how many levels of recursion a user can have in their workflows. From Pegasus’ perspective, recursion in hierarchal workflows ends when a workflow with only compute jobs is encountered . However, the levels of recursion are limited by the system resources consumed by the DAGMan processes that are running (each level of nesting produces another DAGMan process) .
The figure below illustrates an example with recursion 2 levels deep.
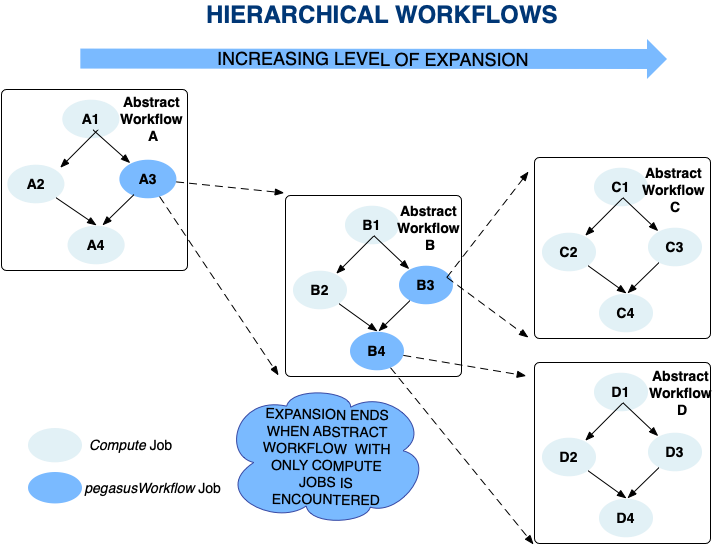
Recursion in Hierarchal Workflows
The execution time-line of the various jobs in the above figure is illustrated below.
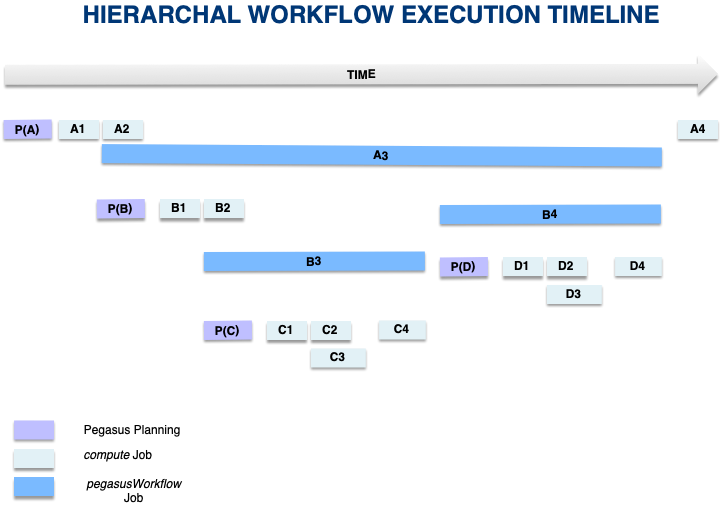
Execution Time-line for Hierarchal Workflows
12.3.5. Hierarchical Workflows with Globus Online Endpoint on Submit Host
If your workflows are using Globus Online for data transfers; then you might find yourself setting up Globus Online endpoint for the submit host (local site) and also describing your Site Catalog accordingly.
x-pegasus: {apiLang: python, createdBy: bamboo, createdOn: '05-01-24T16:11:43Z'}
pegasus: 5.0.4
sites:
- name: local
arch: x86_64
os.type: linux
os.release: rhel
os.version: '7'
directories:
- type: sharedScratch
path: /local-site/scratch
sharedFileSystem: true
fileServers:
- {url: 'go://565ssssxxxx/local-site/scratch', operation: all}
However, this configuration can cause errors with hierarchical workflows, with the prescript for the sub workflow failing since it tried to do a transfer between GO endpoint and a file URL; which is not supported by Globus Online.
To get around this, you need to ensure * the sharedFileSystem attribute for the sharedScratch directory for the
local site is set to True, as in the snippet above.
if you have a compute job in the workflow that generates the abstract workflow for
12.4. Optimizing Data Transfers
Issue: When it comes to data transfers, Pegasus ships with a default configuration which is trying to strike a balance between performance and aggressiveness. We obviously want data transfers to be as quick as possibly, but we also do not want our transfers to overwhelm data services and systems.
Solution: Starting 4.8.0 release, the default configuration of Pegasus now adds transfer jobs and cleanup jobs based on the number of jobs at a particular level of the workflow. For example, for every 10 compute jobs on a level of a workflow, one data transfer job( stage-in and stage-out) is created. The default configuration also sets how many threads such a pegasus-transfer job can spawn. Cleanup jobs are similarly constructed with an internal ratio of 5.
Additionally, Pegasus makes use of DAGMan categories and associates the following default values with the transfer and cleanup jobs.
See Job Throttling for details on how to set these values.
Information on how to control manully the maxinum number of stagein and stageout jobs can be found in the Data Movement Nodes section.
To control the number of threads pegasus-transfer can use in standard transfer jobs and when invoked by PegasusLite, see the pegasus.transfer.threads property.
12.5. Job Throttling
Issue: For large workflows you may want to control the number of jobs released by DAGMan in local condor queue, or number of remote jobs submitted.
Solution: HTCondor DAGMan has knobs that can be tuned at a per workflow level to control it’s behavior. These knobs control how it interacts with the local HTCondor Schedd to which it submits jobs that are ready to run in a particular DAG. These knobs are exposed asDAGMan profiles (maxidle, maxjobs, maxpre and maxpost) that you can set in your properties files.
Within a single workflow, you can also control the number of jobs submitted per type ( or category ) of jobs. To associate categories, you needs to associate dagman profile key named category with the jobs and specify the property dagman.[CATEGORY-NAME].* in the properties file. More information about HTCondor DAGMan categories can be found in the HTCondor Documentation.
By default, pegasus associates default category names to following types of auxillary jobs
DAGMan Category Name |
Auxillary Job applied to. |
Default Value Assigned in generated DAG file |
stage-in |
data stage-in jobs |
10 |
stage-out |
data stage-out jobs |
10 |
stage-inter |
inter site data transfer jobs |
|
cleanup |
data cleanup jobs |
4 |
registration |
registration jobs |
1 (for file based RC) |
Below is a sample properties file that illustrates how categories can be specified in the properties file
# pegasus properties file snippet illustrating
# how to specify dagman categories for different types of jobs
dagman.stage-in.maxjobs 4
dagman.stage-out.maxjobs 1
dagman.cleanup.maxjobs 2
HTCondor also exposes useful configuration parameters that can be specified in it’s configuration file (condor_config_val -conf will list the condor configuration files), to control job submission across workflows. Some of the useful parameters that you may want to tune are
HTCondor Configuration Parameter |
Description |
|---|---|
Parameter:START_LOCAL_UNIVERSE
Sample Value :TotalLocalJobsRunning < 20
|
Most of the pegauss added auxillary jobs (createdir, cleanup, registration
and data cleanup ) run in the local universe on the submit host. If you
have a lot of workflows running, HTCondor may try to start too many
local universe jobs, that may bring down your submit host. This global
parameter is used to configure condor to not launch too many local
universe jobs.
|
Parameter:
GRIDMANAGER_MAX_JOBMANAGERS_PER_RESOURCE
Sample Value :Integer
|
For grid jobs of type gt2, limits the number of globus-job-manager
processes that the condor_gridmanager lets run at a time on the
remote head node. Allowing too many globus-job-managers to run
causes severe load on the head note, possibly making it non-functional.
Usually the default value in htcondor ( as of version 8.3.5) is 10.
This parameter is useful when you are doing remote job submissions
using HTCondor-G.
|
Parameter:
GRIDMANAGER_MAX_SUBMITTED_JOBS_PER_RESOURCE
Sample Value : Integer
|
An integer value that limits the number of jobs that a
condor_gridmanager daemon will submit to a resource.
A comma-separated list of pairs that follows this integer limit will
specify limits for specific remote resources.
Each pair is a host name and the job limit for that host. Consider
the example
GRIDMANAGER_MAX_SUBMITTED_JOBS_PER_RESOURCE = 200, foo.edu, 50, bar.com, 100.
In this example, all resources have a job limit of 200, except foo.edu,
which has a limit of 50, and bar.com, which has a limit of 100. Limits
specific to grid types can be set by appending the name of the grid type
to the configuration variable name, as the example
GRIDMANAGER_MAX_SUBMITTED_JOBS_PER_RESOURCE_CREAM = 300
In this example, the job limit for all CREAM resources is 300.
Defaults to 1000 ( as of version 8.3.5).
This parameter is useful when you are doing remote job submissions
using HTCondor-G.
|
12.5.1. Job Throttling Across Workflows
Issue: DAGMan throttling knobs are per workflow, and don’t work across workflows. Is there any way to control different types of jobs run at a time across workflows?
Solution: While not possible in all cases, it is possible to throttle different types of jobs across workflows if you configure the jobs to run in vanilla universe by leverage HTCondor concurrency limits. Most of the Pegasus generated jobs ( data transfer jobs and auxillary jobs such as create dir, cleanup and registration) execute in local universe where concurrency limits don’t work. To use this you need to do the following
Get the local universe jobs to run locally in vanilla universe. You can do this by associating condor profiles universe and requirements in the site catalog for local site or individually in the transformation catalog for each pegasus executable. Here is an example local site catalog entry.
<site handle="local" arch="x86_64" os="LINUX"> <directory type="shared-scratch" path="/shared-scratch/local"> <file-server operation="all" url="file:///shared-scratch/local"/> </directory> <directory type="local-storage" path="/storage/local"> <file-server operation="all" url="file:///storage/local"/> </directory> <!-- keys to make jobs scheduled to local site run on local site in vanilla universe --> <profile namespace="condor" key="universe">vanilla</profile> <profile namespace="condor" key="requirements">(Machine=="submit.example.com")</profile> </site>Replace the Machine value in requirements with the hostname of your submit host.
Copy condor_config.pegasus file from share/pegasus/htcondor directory to your condor config.d directory.
Starting Pegasus 4.5.1 release, the following values for concurrency limits can be associated with different types of jobs Pegasus creates. To enable the generation of concurrency limits with the jobs set the following property in your properties file.
pegasus.condor.concurrency.limits true
Pegasus Job Type |
HTCondor Concurrency Limit Compatible with distributed condor_config.pegasus |
Data Stagein Job |
pegasus_transfer.stagein |
Data Stageout Job |
pegasus_transfer.stageout |
Inter Site Data Transfer Job |
pegasus_transfer.inter |
Worker Pacakge Staging Job |
pegasus_transfer.worker |
Create Directory Job |
pegasus_auxillary.createdir |
Data Cleanup Job |
pegasus_auxillary.cleanup |
Replica Registration Job |
pegasus_auxillary.registration |
Set XBit Job |
pegasus_auxillary.chmod |
User Compute Job |
pegasus_compute |
Note
It is not recommended to set limit for compute jobs unless you know what you are doing.
12.6. Increase Memory Requirements for Retries
Issue: Setting memory limits for codes with varying amounts of memory requirments can be challenging. Some codes do not use much RAM most of the time, but once in a while require more RAM due to for example initial condition and hitting a particular spot in the algorithm.
Solution: A common approach is to provide a smaller limit for the first try of a job, and if the job fails, increase the limit for subsequent tries. This can be accomplished with an expression for the request_memory attribute. For example, setting the attribute in the site catalog, setting the limit to 1 GB for the first try, and then 4 GB for remaining tries:
<profile namespace="condor" key="request_memory"> ifthenelse(isundefined(DAGNodeRetry) || DAGNodeRetry == 0, 1024, 4096) </profile>
12.7. Slot Partitioning and CPU Affinity in Condor
By default, Condor will evenly divide the resources in a machine (such as RAM, swap space and disk space) among all the CPUs, and advertise each CPU as its own slot with an even share of the system resources. If you want to have your custom configuration, you can use the following setting to define the maximum number of different slot types:
MAX_SLOT_TYPES = 2
For each slot type, you can divide system resources unevenly among your CPUs. The N in the name of the macro listed below must be an integer from 1 to MAX_SLOT_TYPES (defined above).
SLOT_TYPE_1 = cpus=2, ram=50%, swap=1/4, disk=1/4
SLOT_TYPE_N = cpus=1, ram=20%, swap=1/4, disk=1/8
Slots can also be partitioned to accommodate actual needs by accepted jobs. A partitionable slot is always unclaimed and dynamically splitted when jobs are started. Slot partitioning can be enable as follows:
SLOT_TYPE_1_PARTITIONABLE = True
SLOT_TYPE_N_PARTITIONABLE = True
Condor can also bind cores to each slot through CPU affinity:
ENFORCE_CPU_AFFINITY = True
SLOT1_CPU_AFFINITY=0,2
SLOT2_CPU_AFFINITY=1,3
Note that CPU numbers may vary from machines. Thus you need to verify what is the association for your machine. One way to accomplish this is by using the lscpu command line tool. For instance, the output provided from this tool may look like:
NUMA node0 CPU(s): 0,2,4,6,8,10
NUMA node1 CPU(s): 1,3,5,7,9,11
The following example assumes a machine with 2 sockets and 6 cores per socket, where even cores belong to socket 1 and odd cores to socket 2:
NUM_SLOTS_TYPE_1 = 1
NUM_SLOTS_TYPE_2 = 1
SLOT_TYPE_1_PARTITIONABLE = True
SLOT_TYPE_2_PARTITIONABLE = True
SLOT_TYPE_1 = cpus=6
SLOT_TYPE_2 = cpus=6
ENFORCE_CPU_AFFINITY = True
SLOT1_CPU_AFFINITY=0,2,4,6,8,10
SLOT2_CPU_AFFINITY=1,3,5,7,9,11
Please read the Condor Administrator’s Manual for full details.