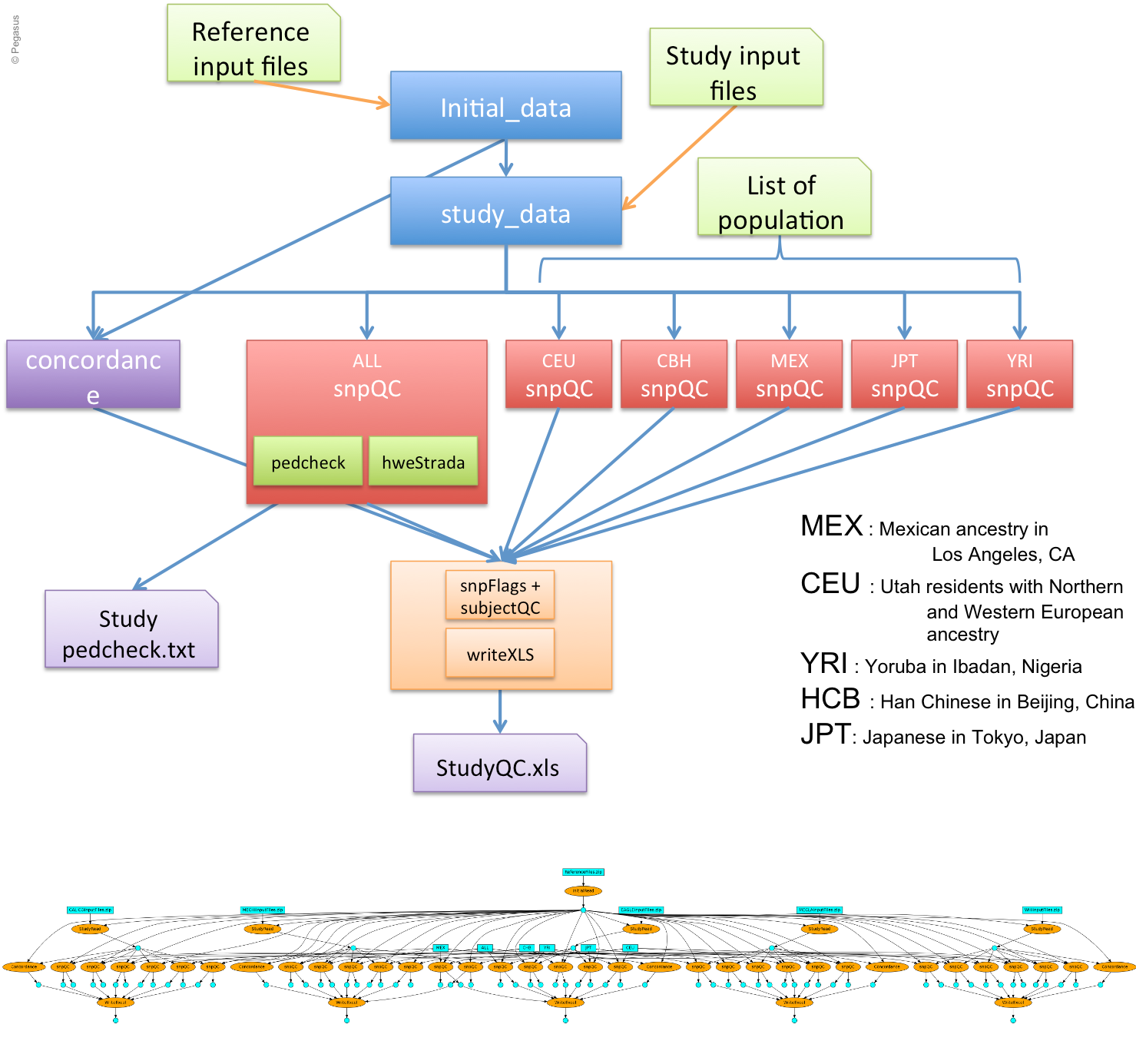
This workflow aims to indicate discrepancy in the data coming from different group and checks of concordance on the genotype calls against HapMap genotypes. Initially, it was a big R scripts which computed all the steps at once, sequentially. We have split the code into different steps/tasks to represent them as workflow. As a result, we manage to execute the whole workflow within 30 min on a small cluster compare to the 4 hours of the initial sequential version.
Scientists:Steve Buyske (Rutgers University)