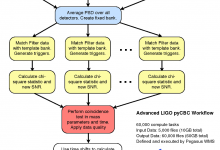
Advanced LIGO - Laser Interferometer Gravitational Wave Observatory
The Pegasus team is very happy to hear about LIGO’s incredible discovery: the first detection of gravitational waves from colliding black holes. We congratulate the entire LIGO Scientific Collaboration and the Virgo Collaboration on this incredible achievement. The Pegasus team is very pleased to have contributed to LIGO’s software infrastructure. One … Read More

Event Horizon Telescope
The Event Horizon Telescope (EHT) achieved the first horizon-scale image of a black hole by linking radio telescopes worldwide, forming a high-resolution, virtual telescope. This global collaboration of about 300 members uses advanced computational workflows, relying on Pegasus and the OSG OSPool to process vast data from EHT observations. Synthetic … Read More

Southern California Earthquake Center - CyberShake and Broadband
Southern California Earthquake Center (SCEC) is a community of over 600 scientists, students, and others at over 60 institutions worldwide, headquartered at the University of Southern California. SCEC is funded by the National Science Foundation and the U.S. Geological Survey to develop a comprehensive understanding of earthquakes in Southern California … Read More

XENONnT - Dark Matter Search
The XENON collaboration has utilized the Pegasus workflow management system for a number of years for their Monte Carlo simulations. In XENON1T, these simulations allowed for a very careful characterization of the expected backgrounds and played a crucial role in the interpretation of the experimental results, both for the 2nDEC … Read More
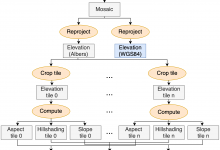
SOil MOisture SPatial Inference Engine (SOMOSPIE)
SOMOSPIE is an advanced earth science engine utilizing machine learning (ML) models to generate high-resolution soil moisture predictions from 27km-resolution satellite data. By integrating data from the ESA-CCI soil moisture database with hydrologically relevant terrain parameters for targeted regions, SOMOSPIE performs effective downscaling. This approach represents an alternative to traditional … Read More

Structural Protein-Ligand Interactome (SPLINTER)
The Structural Protein-Ligand Interactome (SPLINTER) project predicts the interaction of thousands of small molecules with thousands of proteins. These interactions are predicted using the three-dimensional structure of the bound complex between each pair of protein and compound that is predicted by molecular docking. These docking runs consist of millions of … Read More
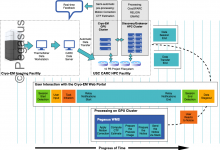
CryoEM Workflows at USC
In 2022, USC New Cryogenic Electron Microscopy Facility officially opened for business. The facility houses two state-of-the-art electron microscopes capable of imaging molecules. CryoEM mainly focuses on structural biology and centers on studying the shapes of biological components, such as proteins and ribosomes, and how their shapes change as they … Read More
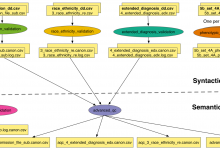
NRGR - Quality Control
The NIMH Center for Collaborative Genomic Studies on Mental Disorders was established through the NIMH Human Genetics Initiative in 1998 to leverage and increase the value of human genetic samples and data produced through NIMH funded research. NIMH Repository and Genomics Resource (NIMH-RGR) plays a key role in facilitating psychiatric … Read More
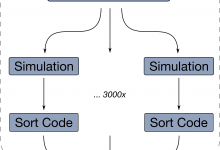
Investigating the Strong Nuclear Force
In general if you want to measure something very small, you need a very large microscope; if you want to measure the nuclear strong force, the force that holds the nuclei of an atom together, you need a very large microscope. One such microscope is the Gamma Ray Infrastructure For … Read More
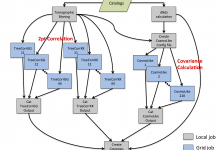
WLPipe - Weak Gravitational Lensing Pipeline
One of the most exciting and challenging areas of modern cosmology is weak gravitational lensing: the phenomenon of small distortions in the shapes of background galaxies as the light they emit traverses the lumpy universe. By measuring the shapes of galaxies in a given region, cosmologists can infer the total … Read More
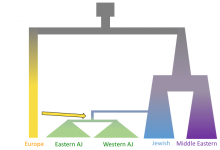
Inference of human demographic history with Approximate Bayesian Computation
This project focuses on human population genomics. We infer human demographic history, such as global migrations, population size changes, and mixing between populations through modeling. This type of research satisfies an innate human interest to understand our own history, and provides a foundation for medical genomics research. Specifically, in this … Read More
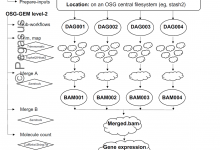
Mining Complex Gene Expression Across the Tree of Life
Modern high throughput DNA sequencing technology continues to revolutionize life science research. However, tens to hundreds of millions of DNA sequence records within tens of thousands of datasets aggregates into petabytes of data. HPC/HTC systems like The Open Science Grid are required to process all this data into useful data … Read More
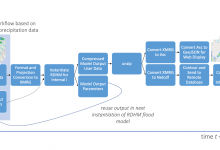
Research Distributed Hydrologic Model
A collaborative effort of several National Oceanic and Atmospheric Administration (NOAA) and National Weather Service (NWS)hydrology research laboratories, the Research Distributed Hydrologic Model (RDHM) (Koren et al. 2004, Reed et al. 2007), was developed to improve streamflow predictions in streams and rivers and to improve flash flood forecasting by incorporating … Read More

Astronomical Image Processing
The Data Intensive Research in Astrophysics and Cosmology institute (DIRAC), University of Washington (WA) Astronomy Department, Legacy Survey of Space and Time (LSST) and Amazon Web Services (AWS) joined forces to develop a proof-of-concept (PoC) leveraging cloud resources to processing astronomical images at scale. The AWS PoC’s goal was to … Read More
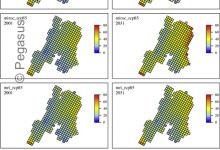
Integrated Assessment Models
Integrated assessment models (IAMs) are commonly used to explore the interactions between different modeled components of socio-environmental systems (SES). They are particularly popular in climate change impacts studies in which climate models are linked to terrestrial process models such as hydrological or lake models to determine impacts of changes in … Read More
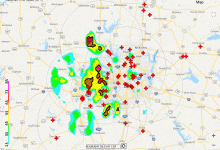
Collaborative and Adaptive Sensing of the Atmosphere
The Collaborative and Adaptive Sensing of the Atmosphere (CASA), has the goal to improve our ability to observe, understand, predict, and respond to hazardous weather events. CASA presents data movement challenges and the need to elastically scale resources on demand. Moment data from a network of seven weather radars, located … Read More
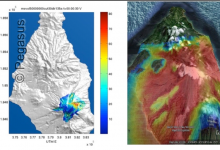
Workflows for Volcanic Mass Flows
Probabilistic hazard risk estimates using Bayesian uncertainty quantification need ensemble methods to explore the parameter space, requiring complex sequences, interactions and exchanges of data and run-time determined computations. In order to make this analysis easier for end researchers to access, a portal based access to the toolkit has been developed. … Read More
Predicting Flash Floods in the Dallas-Fort Worth Metroplex
A collaborative effort of several National Oceanic and Atmospheric Administration (NOAA) and National Weather Service (NWS)hydrology research laboratories, the Research Distributed Hydrologic Model (RDHM) (Koren et al. 2004, Reed et al. 2007), was developed to improve streamflow predictions in streams and rivers and to improve flash flood forecasting by incorporating … Read More
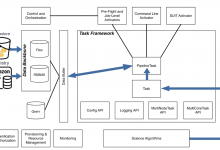
Using the Cloud for Astronomical Image Processing
Overview In 2020 the Data Intensive Research in Astrophysics and Cosmology institute (DIRAC), University of Washington (WA) Astronomy Department, Legacy Survey of Space and Time (LSST) and Amazon Web Services (AWS) joined forces to develop a proof-of-concept (PoC) leveraging cloud resources to processing astronomical images at scale. The AWS PoC’s … Read More
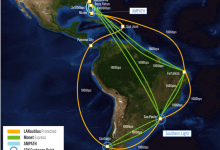
AtlanticWave-SDX and Pegasus
Many new technologies and new paradigms are emerging in the research community to support science workflows, including the use of Data Transfer Nodes (DTNs), distributed compute and data infrastructure, research testbeds, and new inter-domain federated orchestrators. Workflow management systems (WMS), like Pegasus, are evolving to be more resource aware in … Read More
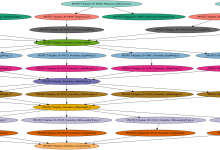
Diffusion Image Processing and Analysis
DIPA is ambitiously meant to be a pipedream for processing and analyzing diffusion weighted magnetic resonance imaging data. The processing involves various machine learning based signal distortion corrections, biophysical diffusion model estimation using non-linear optimization techniques, quality control, image registration using differential geometry both for transformation models and interpolation, region … Read More
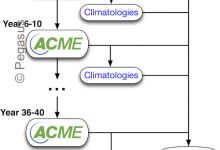
Accelerated Climate Modeling Energy (ACME)
The Accelerated Climate Modeling for Energy (ACME) project is using coupled models of ocean, land, atmosphere and ice to study the complex interaction between climate change and societal energy requirements. One of the flagship workflows of this effort is the fully-coupled climate model running at high resolution. The ACME climate … Read More
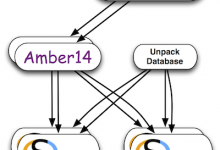
Spallation Neutron Source (SNS)
The Spallation Neutron Source (SNS) is a DOE research facility at Oak Ridge National Laboratory that provides pulsed neutron beams for scientific and industrial research. SNS uses a particle accelerator to impact a mercury-filled target with short proton pulses to produce neutrons by the process of spallation. A wide variety … Read More

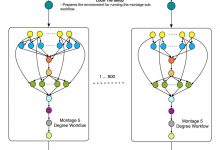
Montage
Caltech astronomers are using Pegasus to generate science-grade mosaics of the sky (Montage project http://montage.ipac.caltech.edu/). Montage delivers science-grade mosaics of the sky. Our technologies were used to transform a single-processor Montage code into a complex workflow and parallelized computations to process larger-scale images. Montage workflows mapped by Pegasus to the … Read More
Galactic Plane
A collaboration with NASA/IPAC Infrared Science Archive (http://irsa.ipac.caltech.edu). The imaging capabilities of the Spitzer Space Telescope have enabled for the first time surveys of the plane of our Galaxy across the infrared spectrum. When taken together with images from existing all sky surveys, these new image surveys contain over 18 … Read More

GT-FAR - Genome and Transcriptome Free Analysis of RNA
GT-FAR is a RNA seq pipeline that allows users to do Alignment, Quantification, Differential Expression, and Variant Calling. This pipeline has been modeled as a Pegasus workflow. Pegasus enables users to execute the pipeline on wide variety of execution environments ranging from local clusters, grids to computational clouds. With funding … Read More
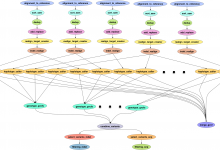
Soybean Knowledge Base (SoyKB) Pipeline
With the advances in next generation sequencing (NGS) technology and significant reduction in sequencing costs it is now possible to sequence large sets of crop germplasm and generate whole genome scale structural variations and genotypic data. In depth informatics analysis of the genotypic data can provide better understanding of … Read More
Genomic Studies of Mental Disorders
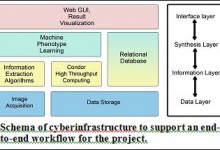
The computational portal developed for the Center for Genomic Studies of Mental Disorders uses Pegasus to manage workflows for genetic population studies. This portal uses the Wings (url) workflow composition system and Pegasus to enable scientists to launch an analysis based on the available workflow template. Below is a screenshot … Read More
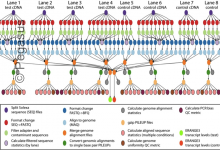
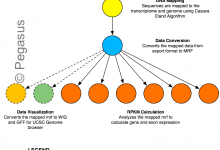
Epigenomics
This application splits sequence files into multiple parts and converts them to the appropriate file format. Then it filters out noisy and contaminating sequences to maps them to their genomic locations. From the individual mapping steps, it merges them into a single global map and uses sequence maps to calculate … Read More
Plant Development
Plant scientists at University of Wisconsin Madison are using Pegasus to generate movies of plant root growth and analyze images collected via time-lapse photography. Another project samples forest locations to characterize the understory vegetation to determine how different plant species are distributed in the woods. Edgar Spalding, a Professor of … Read More
Periodograms
The periodogram application processes time-series data collected by NASA’s Kepler mission. The Kepler satellite uses high-precision photometry to search for exoplanets transiting their host stars. In 2009 the Kepler mission began a multi-year transit survey of 170,000 stars near the constellation Cygnus. In 2010 the project released a data set … Read More
OpenSees Workflows on NEESHub
Over the past several years, the US National Science Foundation has been funding the development of collaborative web sites or ‘collaboratories’. Many communities have adopted the HUBzero platform to create collaboratories called ‘hubs’ where they can share ideas, models, experiences, publications, and data in pursuit of research and education. Hubs … Read More

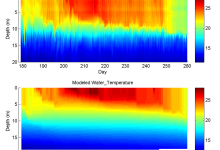
Ocean Forecast
Researchers at the Jet Propulsion Laboratory are exploring Pegasus WMS to run ocean forecast ensembles of the California coastal region. These models produce a number of daily forecasts for water temperature, salinity, and other measures. The main forecast workflow (shown below) consumes about 1.8Gb Data and produces output of about … Read More
Ecosystem Modeling
Professor Paul Hanson at the Center for Limnology at UW Madison conducts research in carbon cycling, ecosystem variability, microbes, algae, and even fish. Limnology is the study of inland water bodies. Large coupled hydrodynamic and biological models have proven to be a powerful tool to help understand the complex dynamics … Read More
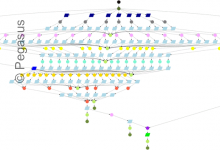
DNA Sequencing
The USC Epigenome Center is currently using the Illumina Genetic Analyzer (GA) system to generate high throughput DNA sequence data (up to 8 billion nucleotides per week) to map the epigenetic state of human cells on a genome-wide scale. Epigenomic Workflow (computational jobs are shown as circles, data transfer jobs … Read More

Computer Vision
Li Zhang is faculty in the UW Madison Computer Sciences Department, and has interests in computer vision and graphics. Graduate student Alok Deshpande’s project is interdisciplinary research that seeks to realize bio-inspired intelligent micro optical imaging systems. Six types of natural eyes are selected to provide inspirations to meet those … Read More
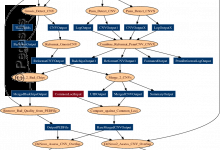
Combined CNV Detection
This workflow combines results from the GNOSIS CNV (CopyNumber Variation) detection algorithm and PennCNV detection algorithm (including X-Chromosome detection) to produce a file containing Merged CNVs. It then processes these further to compare the results with a list of Common CNVs, and assesses the overlap of parent and children CNVs. … Read More
Children’s Hospital of Philadelphia Pediatric Genome Analysis
“The Center for Biomedical Informatics at the Children’s hospital of Philadelphia is the home for the development of innovative solutions to healthcare’s immediate and long-term informatics needs. CBMi provides informatics-focused services, applications, and educational programs to Children’s Hospital clinicians and researchers and seek to transform their craft with high-impact, low-cost … Read More
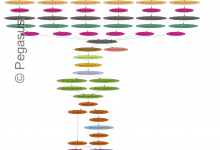
Brain Span
The Brain Span project seeks to find when and where in the brain a gene is expressed. This information holds clues to potential causes of disease. A recent study found that forms of a gene associated with schizophrenia are over-expressed in the fetal brain. To make such discoveries about what … Read More

BioChemistry
George Phillips of the Biochemistry Department at UW Madison has interests in the structure and dynamics of proteins as well as in computational biology. Scientists: George Phillips, UW Madison
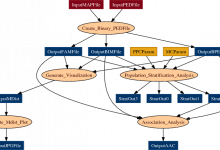
Association Test
This application uses the Cochran-Mantel-Haenszel (CMH) association statistic to do an association test conditional on the matching done in the population stratification step. The clustering uses Plink’s Inheritance-by-Structure (IBS) clustering. Scientists:Christopher Mason, Stephan Sanders, and Matthew State (Yale)
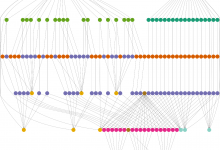
Association Mapping and Population Genetics in Vervets
As the second OWM (old world monkey) sequenced (the first is Rhesus macaque), vervets, unlike the great apes who are mostly in near-extinction status, are widely available for biomedical research. (Rhesus is widely available in India but the export restriction imposed by the Indian government makes it less ideal for … Read More
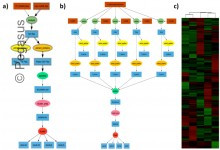
Proteomics
Scientists at OSU use Pegasus for mass-spectrometry-based proteomics. Proteomics workflows have been executed on local clusters and cloud resources. Example proteomic workflow: a) Pegasus workflow template. Square boxes with double lines represent file collections and the ellipses with double boundary represent parallel jobs. b) Implementation of workflow for clustering of … Read More
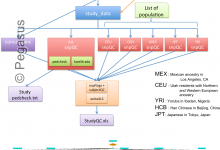
Quality Control of Population Studies
This workflow aims to indicate discrepancy in the data coming from different group and checks of concordance on the genotype calls against HapMap genotypes. Initially, it was a big R scripts which computed all the steps at once, sequentially. We have split the code into different steps/tasks to represent them … Read More
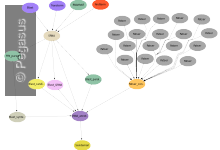
SIPHT
This application is conducting a wide search for small untranslated RNAs (sRNAs) that regulate several processes such as secretion or virulence in bacteria. The kingdom-wide prediction and annotation of sRNA encoding genes involves a variety of individual programs that are executed in the proper order using Pegasus. These involve the … Read More

Solar Dynamics Observatory (SDO)
The Solar Dynamics Observatory (SDO) is NASA’s most important solar physics mission of this coming decade. To be launched near the end of 2008, the three primary instruments on board SDO are the Helioseismic and Magnetic Imager (HMI), the Atmospheric Imaging Assembly (AIA) and the Extreme ultraviolet Variability Experiment (EVE). … Read More